Deciphering the History of ERK Activity from Fixed-Cell Immunofluorescence Measurements
Live-cell ERK biosensor microscopy followed by cyclic immunofluorescence measurements on the same cells.
Abstract
The Ras/ERK pathway drives cell proliferation and other oncogenic behaviors, and quantifying its activity in situ is of high interest in cancer diagnosis and therapy. Pathway activation is often assayed by measuring phosphorylated ERK. However, this form of measurement overlooks dynamic aspects of signaling that can only be observed over time. In this study, we combine a live, single-cell ERK biosensor approach with multiplexed immunofluorescence staining of downstream target proteins to ask how well immunostaining captures the dynamic history of ERK activity. Combining linear regression, machine learning, and differential equation models, we develop an interpretive framework for immunostains, in which Fra-1 and pRb levels imply long term activation of ERK signaling, while Egr-1 and c-Myc indicate recent activation. We show that this framework can distinguish different classes of ERK dynamics within a heterogeneous population, providing a tool for annotating ERK dynamics within fixed tissues.
A dataset linking live-cell ERK activity to ERK target immunofluorescence
To create a dataset that enables correlation of ERK activation to downstream target expression and modification, we first collected live ERK activity measurements in response to differential activation of the RAS/MAP Kinase pathway. We used EKAR 3.5, a calibrated, FRET-based biosensor of ERK activity, to measure single-cell activation in MCF10A mammary epithelial cells. With a series of Epidermal Growth Factor (EGF) concentrations, ERK activity was stimulated in a dose-dependent manner
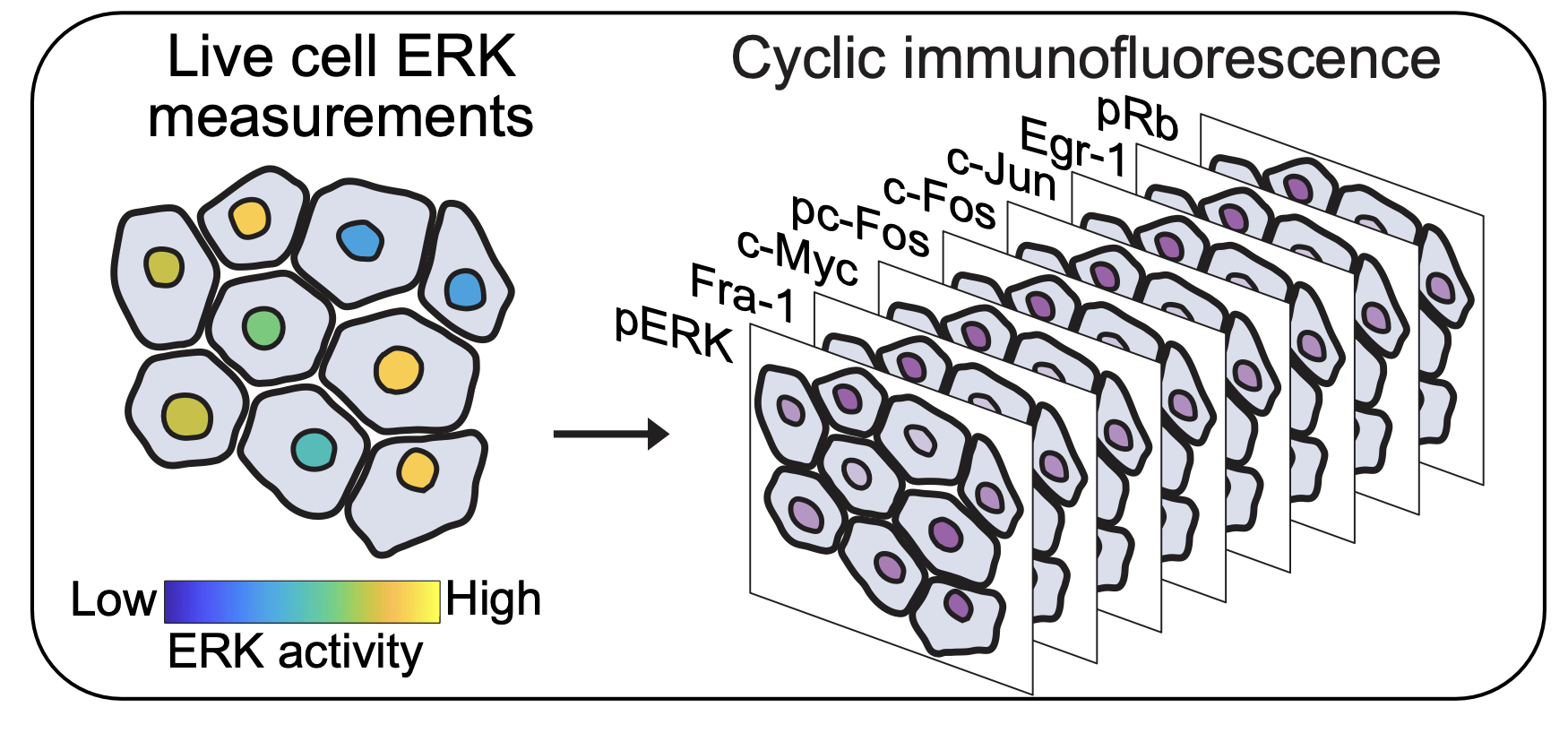
Experimental setup
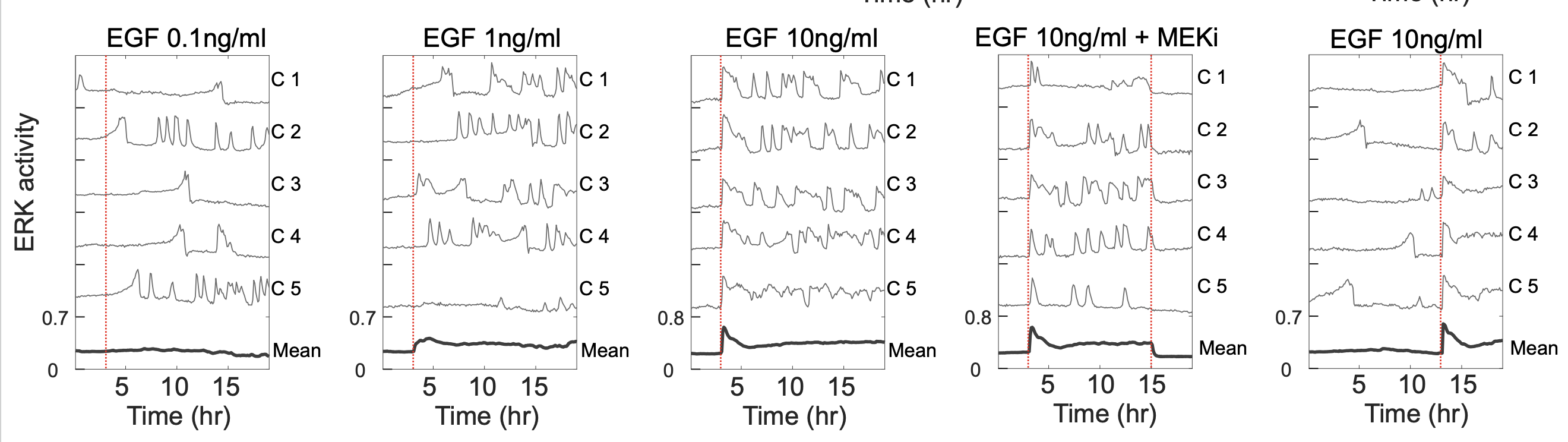
Single-cell ERK actvity measurements.
Immediately following live-cell data collection, we fixed the cells, and conducted cyclic immunofluorescence (4i) staining to measure levels of eight targets downstream of ERK.
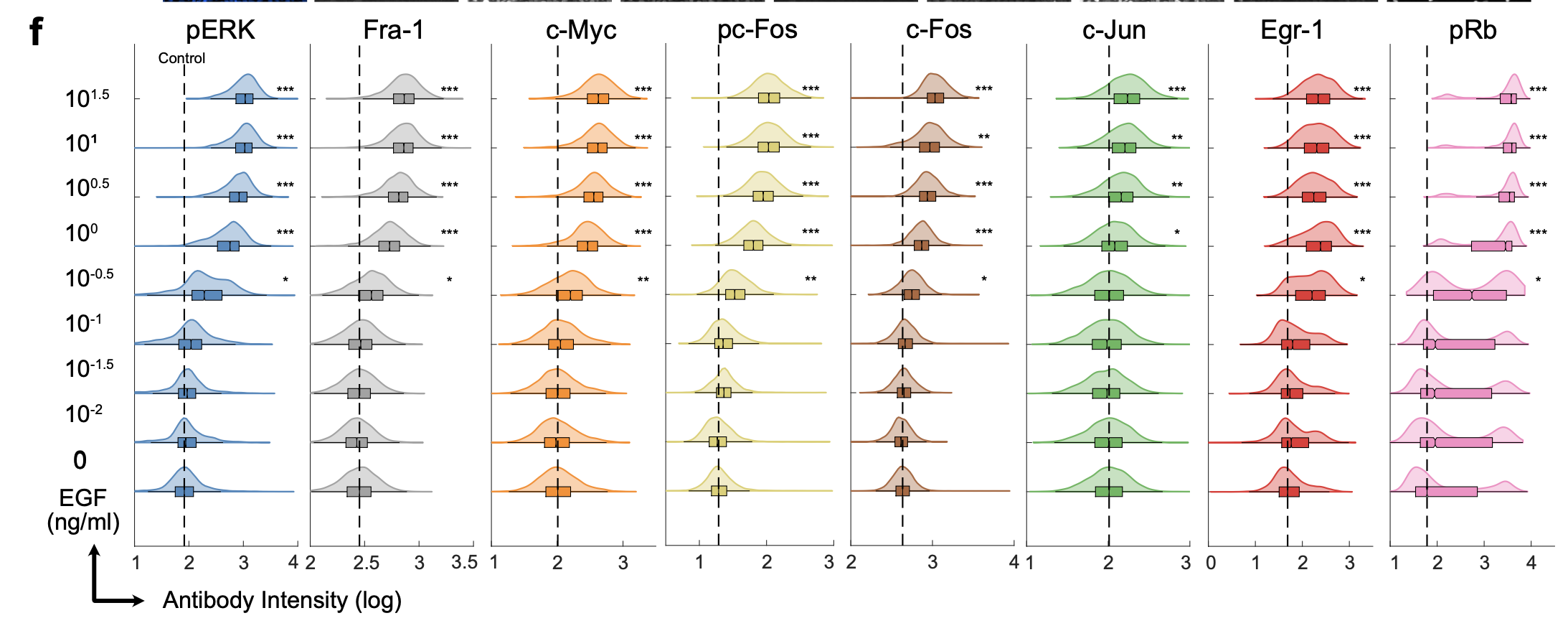
Quantification of ERK Target protein expression via Immunofluorescence.
We then analyzed the correlation between ERK activity and the expression of each target. To link live-cell ERK activity measurements with the respective 4i data for each cell, we aligned the corresponding image datasets and generated a heatmap arranged by the mean ERK activity measurement in each cell. While both ERK activity and 4i targets were variable across the data set, most of the 4i targets exhibited some discernible correlation with mean ERK activity, which was especially strong for Fra-1 and pRb.
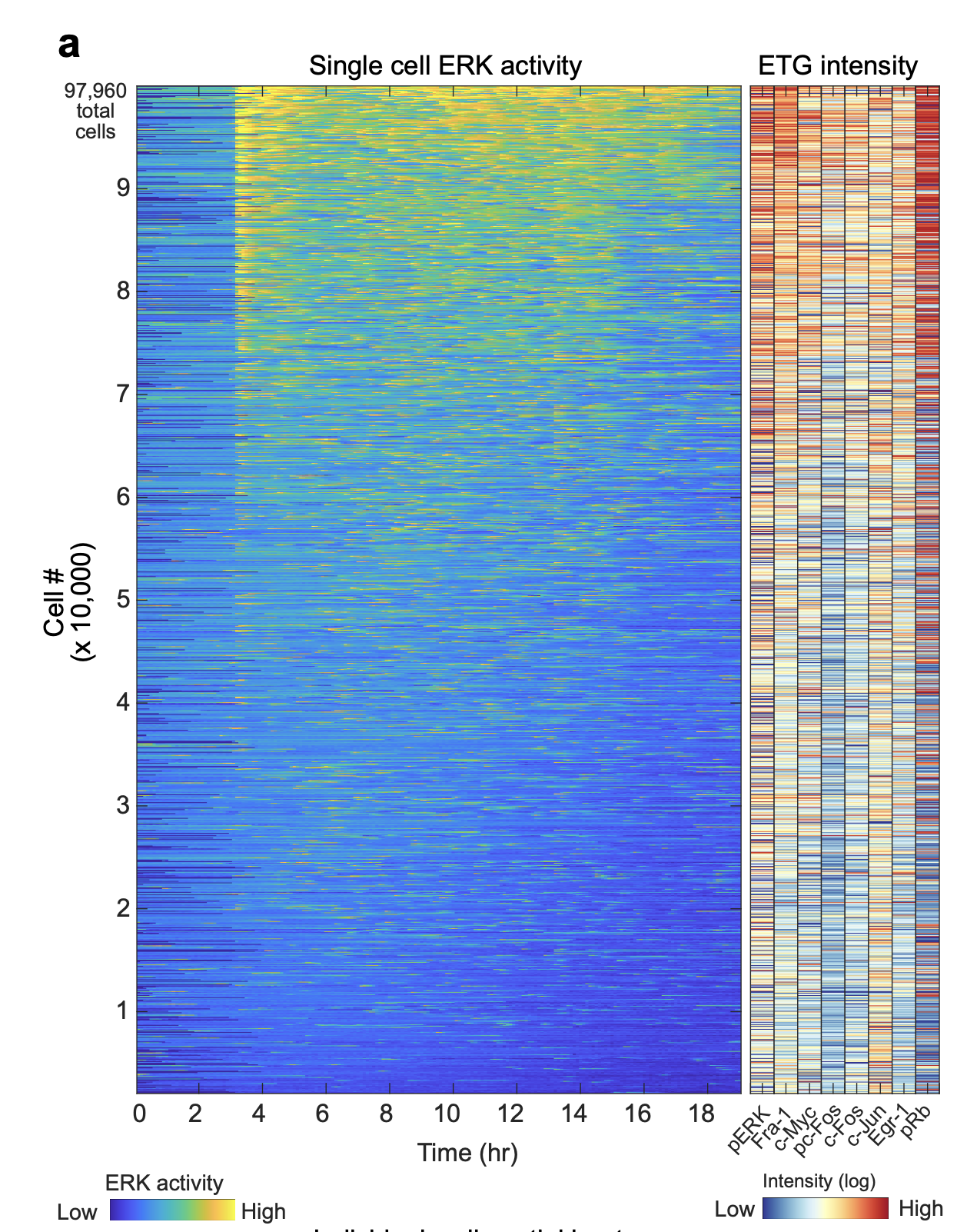
Single-cell heatmap of ERK activity coupled with protein expression.
Regression modeling
For a rigorous statistical analysis of the relationship between ERK activity and ETG expression, we performed cross-validated linear regression using the 4i measurements as predictors and ERK pulse features as response variables. We first created single predictor models to assess how well each target individually predicts each ERK feature in an individual cell. Analysis of the variance explained (R2) for each model confirms the results from the Pearson correlation analysis. Fra-1 and pRb best predict the sum of duration of ERK pulses (R2 = 0.42, 0.31, respectively), and the average ERK activity in each cell (R2 = 0.28 and 0.23). These results suggest the duration of ERK activation seems to have a stronger influence on gene expression than the strength of the activation.
To assess whether the prediction models can be improved by considering multiple stains simultaneously, we generated several multiple linear regression (MLR) models using all 4i measurements as predictors at once. The ERK parameter with the highest variance explained was the sum of duration (R2 = 0.52). The models for derivative benefited the most from the multivariable models, however, they still only explained 33% of the variation in the data. c-Jun again served as a negative control, as those models did not explain any of the variation in ERK activity. We then investigated which antibody combinations are most important in the MLR models. For each ERK feature, we created models that successively added predictors, and measured the resulting R2, and test-set error for each new predictor. For most ERK features, we found that the maximum R2 values can be achieved with just 2 to 3 predictors, where adding Fra-1 and pRb typically caused the highest improvement in R2 values and decrease in test-set error. The best model for the average derivative of ERK had a similar R2 with the model for the ERK mean (R2 = 0.36, 0.33, respectively). The main distinction for the average derivative model is the strong contribution from Egr-1.
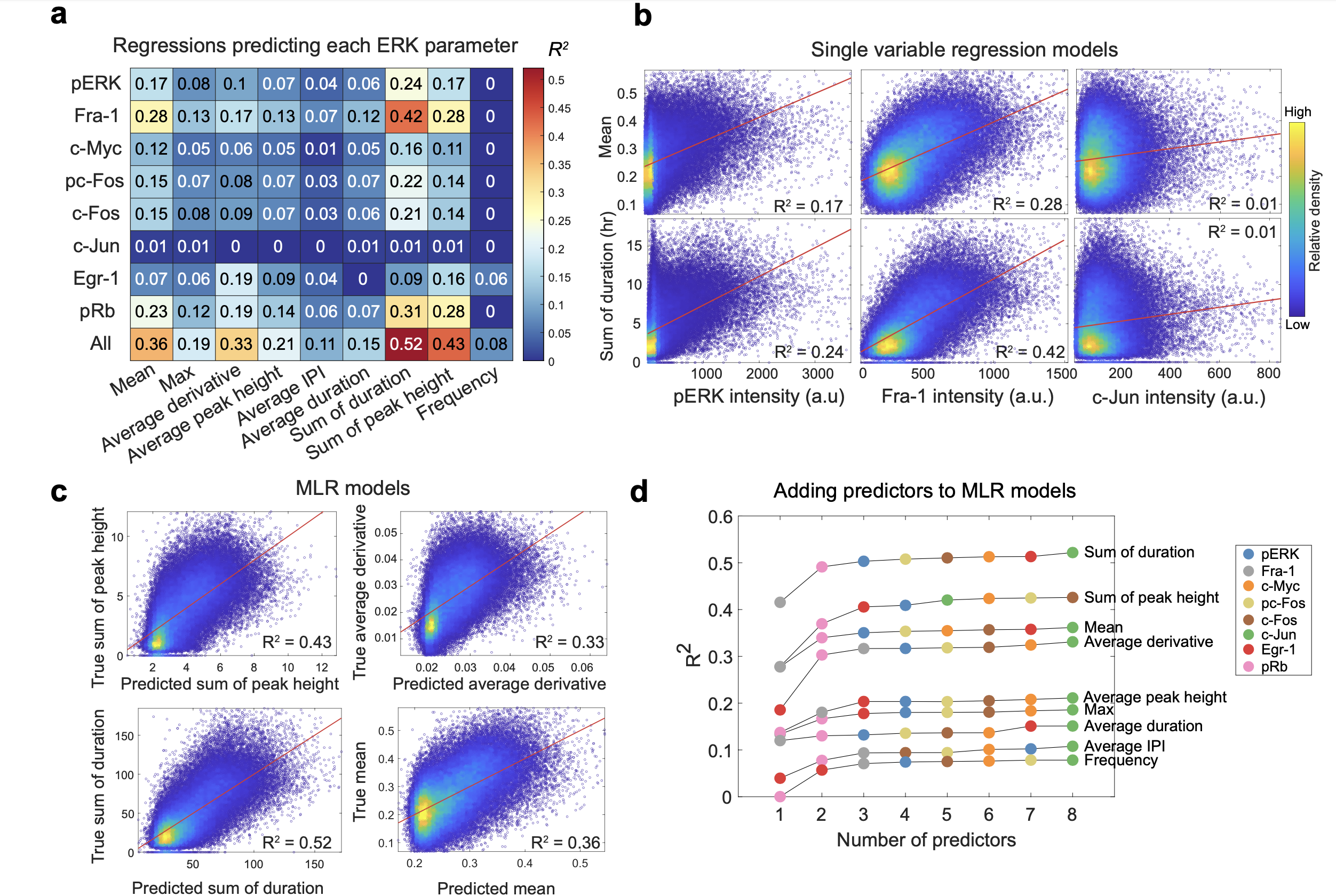
Regression analysis of the relationship between ERK and ETGs
Neural network-based models
While the previous models of featurized ERK activity provide interpretable correlations that help to understand the underlying biological process, they assume linearity and may not capture more complex relationships in the data. Additionally, some ERK parameters are correlated with each other, and other features of the time series may be missed. To examine the importance of the timing of ERK activation and identify which timepoints have he greatest impact on final expression levels, we trained a convolutional neural network (CNN) to use the ERK activity time series to predict expression levels of each ERK target in individual cells. As a comparison to the CNN, we also fit linear regression models (TS linear) using the values at each timepoint of the ERK time series as individual variables to predict final ERK target levels.
We compared the performance of these time series-based models with that of ERK dynamics feature based models (Featurized linear). These feature-based models used all nine featurized ERK measurements (i.e. mean/duration/frequency) to predict the expression of each ERK target. We found that the CNN achieved the highest performance in predicting all ERK targets, except for pERK. To account for overfitting, we calculated the mean squared error (MSE) on unseen data (test set) and the CNN exhibited the least error for all targets, except for pERK. Although the CNN yielded better performance for most targets, a significant amount of variance is still not captured by the model. Notably, the CNN models for Egr-1 and pRb explained much more variance than linear regression models of other targets, implying that Egr-1 and pRb likely respond to ERK activation with significant non-linearity. Finally, for many 4i targets, the featurized linear models underperformed the other two methods, both in R2 and test set error, indicating that the featurization method often does not capture important aspects of ERK signaling that influence gene expression.
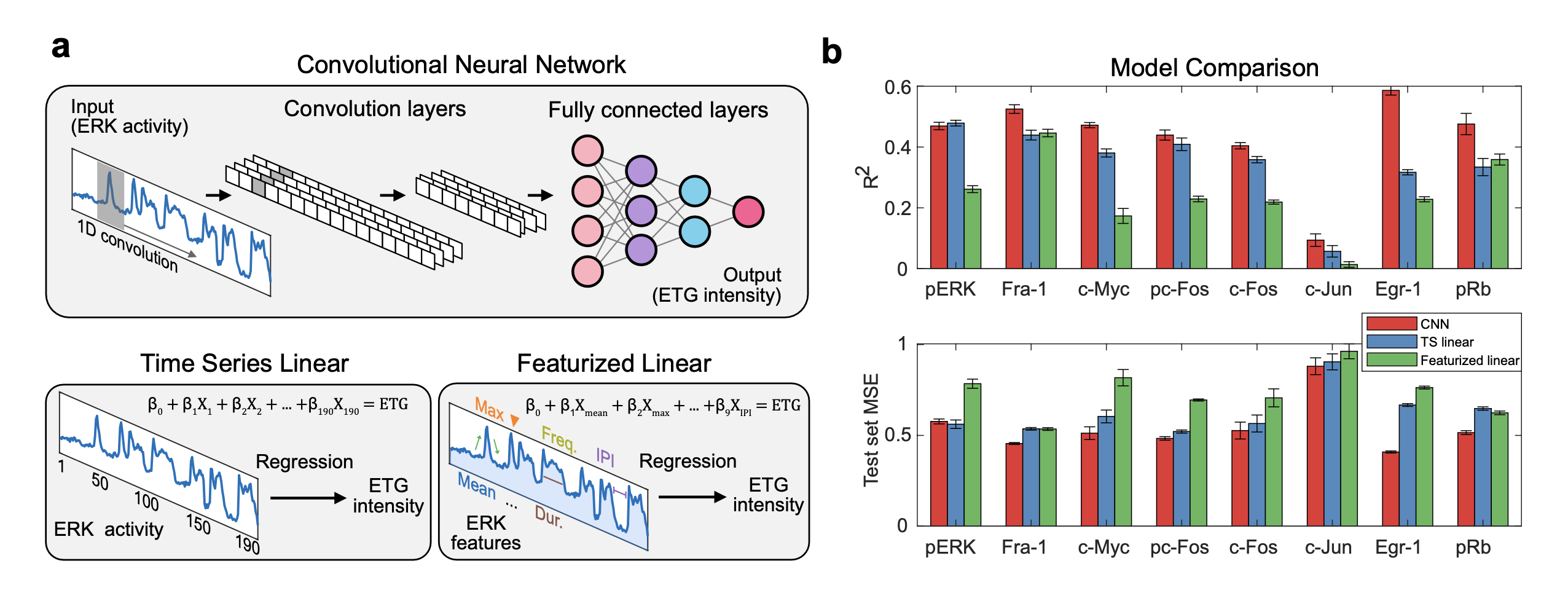
Comparison of CNN and regression models.
Classification models
Thus far, we have trained models that predict several continuous variables that represent ERK history; however, the application of these models is limited by the challenge of concurrently visualizing the predictions. Therefore, we demonstrate here how spatiotemporal ERK predictions can be represented in a concise and intuitive manner. To do so, we first used k-means clustering to group cells into similar response classes, or prototypes, of ERK activity. We clustered cells into five classes: low activation (cluster 1), recent deactivation (cluster 2), long term activation (cluster 3), mid-term activation (cluster 4), and recent activation (cluster 5) Analysis of the 4i target expression levels in each cluster was consistent with our previous statistical models. Long-term activation led to the highest expression of pERK, Fra-1, and pRb, while low activation displayed the lowest for all targets. Cells with recent activation highly expressed Egr-1 and c-Myc.
We next trained a decision tree classifier that predicts prototypes of ERK signaling history using ERK target expression levels. The overall prediction accuracy of our model was 51% (compared to 20% for random selection), while individual class predictions varied in accuracy. Long-term activation class predictions were the most accurate (64%), and mid-term activation classifications were the least accurate (36%). These findings indicate that long term and recent activation result in distinct patterns of the expressed genes we measured, while mid-term activation produces the highest variability in gene expression.
Finally, to simulate a potential use case with fixed tissue samples, we then used our classifier to predict ERK activity classes, and therefore histories, in cells from a single well in our dataset. Our analysis effectively quantifies the distinctiveness in gene expression associated with different ERK signaling prototypes and illustrates the utility of ETG stains in predicting the spatiotemporal signaling history of individual cells.
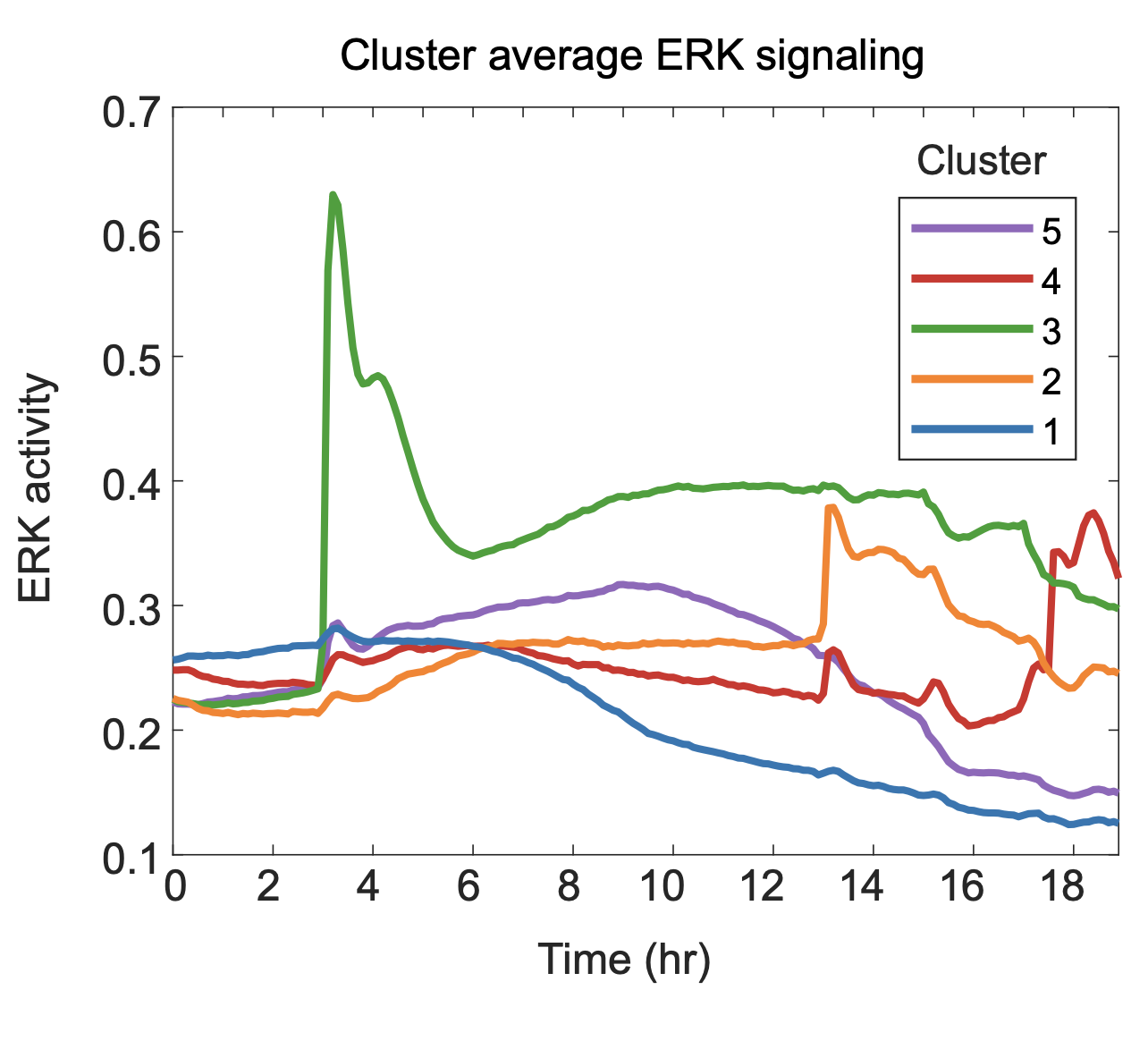
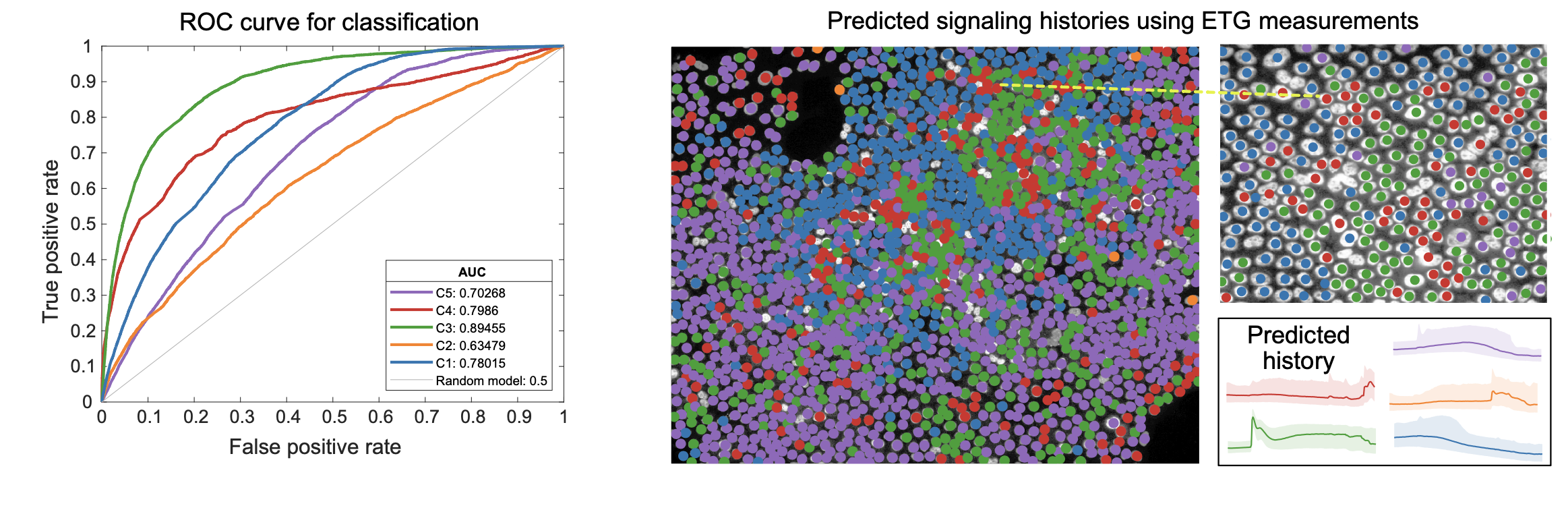
Inferring ERK signaling histories
Systems biology and ODE modeling
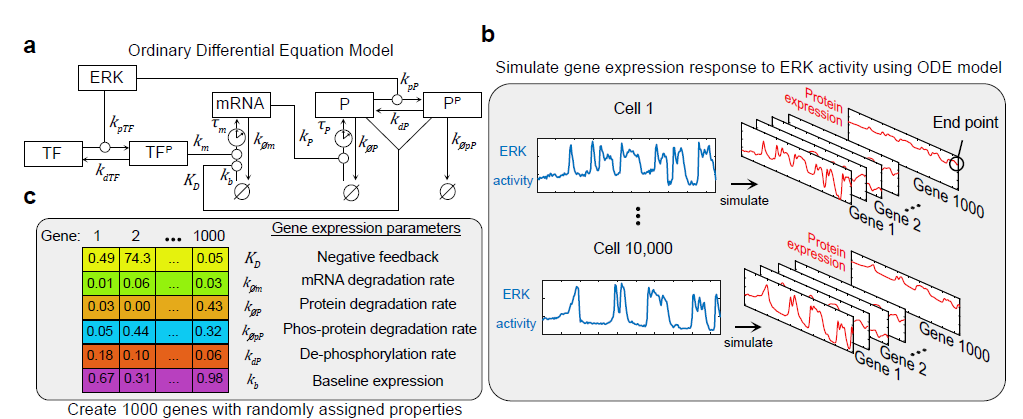
ODE model to simulate expression of 1000 genes
To investigate the theoretical limits of predicting ERK dynamics from ETG levels, we extended an ordinary differential equation (ODE) model representing the regulation of ETGs (a). For a given ERK activity time series, the model simulates the mRNA and protein levels of a hypothetical ERK-responsive gene (sim-ETG). (b) We constructed 1,000 hypothetical sim-ETGs by randomly assigning each one with different parameters values for mRNA degradation rate, protein degradation rate, phosphorylated protein degradation rate, protein dephosphorylation rate, negative feedback half-max concentration, and fractional expression at baseline (Fig. 6c, Supplementary Table 2). These 1,000 gene parameter configurations survey the parameter space with the goal of identifying sim-ETGs that capture different aspects of ERK signaling. Using 10,000 randomly selected live-cell ERK activity measurements from our experimental data, we simulated responses of all 1,000 sim-ETGs for each cell
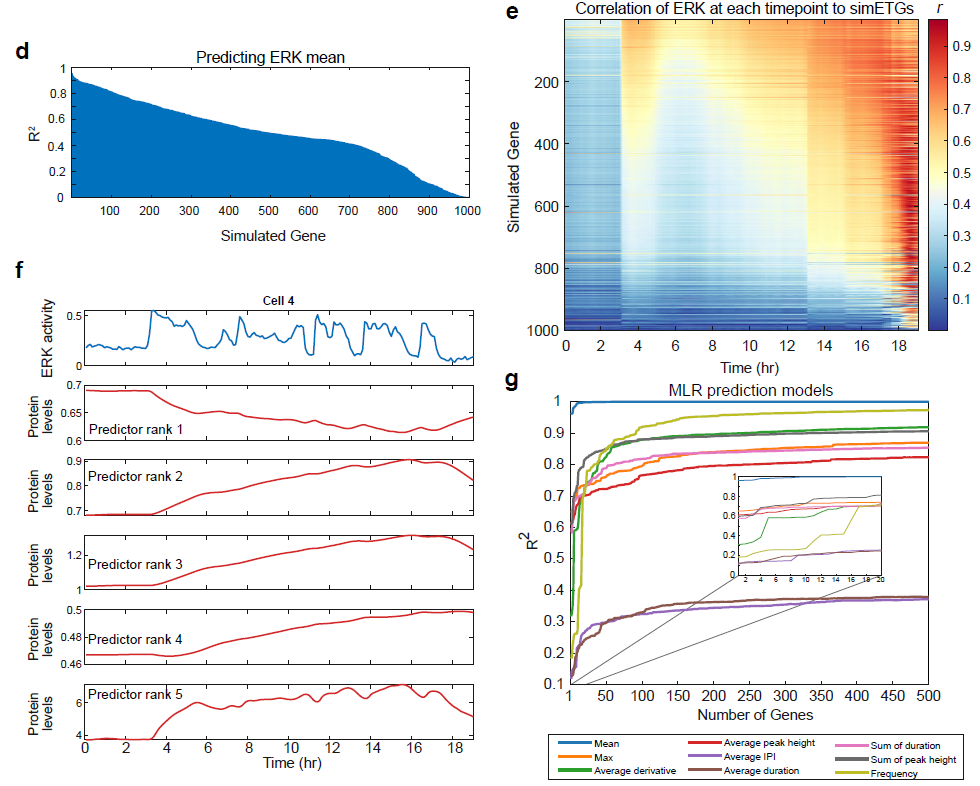
Using the end point sim-ETG protein values (representing a fixed-cell 4i measurement of the hypothetical protein), we applied single variable regression modeling to characterize each sim-ETG’s capacity to predict ERK dynamics features. For predicting average ERK activity throughout the experiment, we found that 49% of sim-ETGs exhibited a R2 above 0.5 and over 100 were excellent predictors (R2 > 0.8) (d). For predicting the maximum activation and average pulse height, only 12% of sim-ETGs exhibited a R2 above 0.5, with a maximum R2 around 0.6.
Models for predicting dynamic ERK features like the frequency or the average derivative were overall worse than integrative features like the mean or sum of duration, reflecting that sim-ETGs under this model are variations on an integrator of ERK activity.. To visualize the gene expression response, we plotted a single cell’s ERK signal along with the response of the top five predictors of the mean (f). These response profiles show that both genes activated by or inhibited by ERK can serve as reliable predictors of ERK activity. While our experimental ETG measurements were selected based on known positive responders to ERK, 20% of sim-ETGs were negatively regulated by ERK; experimental prediction of ERK activity would likely be improved by including genes that are inhibited by ERK (Yamamoto et al., 2006).
We then analyzed which gene parameters most influence how well an individual sim-ETG predicts mean ERK activity by examining the weights from a MLR model of sim-ETGs. Consistent with the known behavior of Fra-1, slow mRNA and phosphorylated protein degradation rates allow for accurate recording of the average ERK history. Our 4i data analysis determined that while Fra-1 predicts long-term history, Egr-1 and c-Myc predict recent history. To examine this distinction in sim-ETGs, we calculated the correlation between the ERK activity at each timepoint and end-point protein expression. As expected, genes that predict mean ERK activity tend to be correlated with ERK activity over a wide time span, similarly to Fra-1. Those that are less effective at predicting mean are correlated with recent activation, behaving more like Egr-1 or c-Myc.. Notably, no sim-ETG under this model was specifically predictive of intermediate timescales of activation (i.e. 5-10 hours prior to fixation).
Finally, to investigate how many gene measurements are required to accurately predict the different aspects of ERK signaling, we created MLR models which used many sim-ETGs at once to predict multiple ERK pulse features. These models greatly improved our predictions, as most explained 75 to 99% of the variance in the dataset (g). Of note, the derivative and frequency model predictions drastically improved as the number of predictors increased. This result was not obtained through overfitting, as the test set error of the models also decreased with more sim-ETGs. For most ERK features, between 16 to 20 sim-ETGs are required for obtaining good models (R2 ~ 0.7) (g inset). From an experimental standpoint, these results demonstrate that predicting dynamic features of ERK is highly feasible, and depends largely on which gene products are measured. From a practical standpoint, measuring for 20 proteins using a multiplex staining protocol is readily achievable. In all, the ODE model indicates that our ERK activation inference method is a feasible solution for fixed tissue analysis, and will benefit from further exploration of potential endogenous gene products to measure.
Discussion
Here, we provide proof of principle that end-point ETG staining can be used to infer key aspects of long-term ERK activity within fixed cell samples. While differences in ETG activation by ERK were previously known, our analysis formalizes these differences and shows how quantitative models can be used to infer ERK’s activity history with single-cell resolution. The ETG measurements in these experiments provide information about two broad types of ERK behavior, long-term and short-term activation. Additionally, our model analysis of simulated ETGs demonstrates that additional measurements could even more finely resolve signaling patterns, such as intermittent pulses. The experimental and biological limits of these predictions remain to be established; however, this model framework can be used to estimate properties of ETGs that would optimally improve the measurement set.